Abstract
We identified a loose upper panicle mutant (lup) from a japonica-type rice variety, Hwacheongbyeo, treated by Ethyl Methane Sulfonate (EMS). The lup mutant displayed an increased distance between spikelets particularly in the first primary branches, and the number of spikelet was reduced. In addition, aborted spikelets in the tip of first primary branches were observed. Besides these morphological changes in the panicle, the lup mutant also displayed overall reduction in culm length, panicle length, grain weight, and tiller number. On the contrary, the chlorophyll content was relatively high in lup mutant in comparison to wild-type plants, and displayed a “stay-green” phenotype even after physiological maturity. Genetic analysis (using F2 population of lup/M.23) revealed that a single recessive gene is involved in the above-mentioned morphological changes in the lup mutant. A candidate genomic region was fine-mapped at an interval of 1.04 Mb flanked by two molecular markers, 18170 and D0052, on the long arm of chromosome 8. In this region, we found a total of 348 mutation points using a slightly modified MutMap method. Based on these results, we expect the candidate genomic region containing a putative LUP gene will provide an important clue in developmental regulation of spikelets and panicle in rice.
-
Key words: Loose upper panicle, Spikelet distance, Spikelet number per panicle, SNPs, MutMap
INTRODUCTION
Rice (
Oryza sativa L.) is the primary source of calories for more than half of the world population (
Khush 2005). However, owing to a gradual decrease in farmland area, the average annual increase in rice production has been decreasing (
He et al. 2010). It has been suggested that the improvement of rice grain yield per unit area is one of the most efficient ways to overcome such constraints (
Cassman et al. 2003).
Plant architecture in cereal crops is generally considered to be a major factor that influences grain yield through the efficient use of solar radiation and optimal partitioning of photosynthesis into organs (
Wang and Li 2008). Among the important plant organs determining grain productivity, panicle is the key organ which is directly involved in grain productivity, therefore, optimization of panicle structure (such as size and shape) has become a priority objective for higher yield in many of breeding programs (
Sakamoto and Matsuoka 2004). In rice, panicle development is a key event to generate a branched structure that is composed of multiple spikelets. It is generally known that the panicle development is often negatively regulated by genetic or environmental factors and leads to whitish or small size of spikelets accompanied with severe reduction in fertility and grain yield (
Bai et al. 2015). However, the molecular mechanisms to control the panicle development have not yet been widely understood. Spikelet is the basic unit of panicle, and consists of multiple florets (each having a lodicule, carpel, and six stamens) that are surrounded by glumes (
Yoshida and Nagato 2011). The abortion of spikelet is usually affected by degeneration of floral organs, and eventually leads to reduction in grain yield. Spikelet degeneration is generally occurred from basal to apical end of the panicle, and defective and/or whitish spikelet is observed as a result (
Li et al. 2009;
Cheng et al. 2011).
Several studies have reported that the development of rice panicle is possibly regulated by common mechanisms that also regulate the tiller formation and elongation. For instance, two genes,
MONOCULM1 (
MOC1) and
LAX PANICLE1 (
LAX1), are known to be involved in these common mechanisms (
Li et al. 2003;
Oikawa et al. 2009). Those rice plants carrying the mutation in one of these genes displayed a reduction either in tiller number or panicle branch number (
Youlin et al. 2014). In addition, it has been reported that near isogenic lines (NILs) carrying mutation in the
IDEAL PLANT ARCHITECTURE1 (
IPA1) genic region displayed reduced tiller number and increased panicle branches (
Jiao et al. 2010). Besides these, several other genes involved in regulation of panicle development also have been identified. For instance,
DENSE AND ERECT PANICLE 1 (
DEP1) was fine mapped from a major dominant quantitative trait locus (QTL) responsible for control of panicle branches (
Huang et al. 2009).
Grain number 1a/Cytokinin oxidase 2 (
Gn1a/
OsCKX2) gene, which encodes a cytokinin oxidase, has been identified from a major QTL responsible for improvement of grain number (
Li et al. 2013). Moreover,
ABERRANT PANICLE ORGANIZATION 1 (
APO1) had been reported as a pivotal gene to regulate primary panicle branch and to control the vascular bundle formation, which related to an improvement of harvest index and grain yield in rice (
Terao et al. 2010).
In this study, we identified a loose upper panicle mutant (lup) from a japonica-type rice variety, Hwacheongbyeo, which is treated by Ethyl Methane Sulfonate (EMS). The aim of this study is to investigate the significant differences between lup mutant and wild type from phenotypic and genetic perspectives. The phenotypic characterization and genetic analysis of lup mutant revealed that the candidate genomic region for LUP gene is located in the long arm of chromosome 8 flanked by two molecular markers, 18170 and D0052, at an interval of 1.04 Mb. In this candidate region, we identified a total of 348 mutation points in 51 genes by using a slightly modified MutMap method. The results from this study will therefore facilitate the positional cloning and functional characterization of the LUP gene.
MATERIALS AND METHODS
Plant materials
The loose upper panicle mutant (lup) line was induced by chemical mutagenesis of the japonica-type rice variety, Hwacheongbyeo, using Ethyl Methane Sulfonate (EMS) treatment. The seeds of the lup mutant were selected from M2 generation and fixed in pure line by repetitive self-crossing. For the genetic analysis and fine mapping of the lup gene, we constructed two F2 populations from the cross between lup mutant and Milyang 23 and another cross between lup mutant and Hwacheongbyeo. Milyang 23 is a tongil-type cultivar from indica/japonica cross. Field plantation, management and agronomic data were collected in summer season of 2014 at experimental farm station of Seoul National University, Suwon, South Korea. Six plants of each parental cultivars were averaged and used as the measurements for each agronomic traits. The segregation of apical aborted spikelet in both F2 populations were counted after heading. Chi-square test was carried out to determine the fitness to ratio of 3:1 in both F2 populations.
Characterization of the agronomic traits
Agronomic traits of the
lup mutant and wild-type plants were observed during various stages of development, and recorded for further analysis using SAS 9.2 (
https://www.sas.com/). Panicle related traits and other non-panicle related agronomic traits mentioned under were used for
lup mutant genotypic analysis.
To determine the content of chlorophyll present in plant leaves, measurement was done on flag leaf at early reproductive stage of development using Minolta Chlorophyll meter, SPAD 502 (Minolta Camera Co., Japan) and designated as SPAD value. Five spots/leaf at 3–5 cm intervals was measured and mean value was recorded as the amount of chlorophyll content.
Number of tillers and plant height
Tiller number
The tillers number per hill for both lup mutant and wild type was measured by counting the number of tiller from randomly selected six plants and the mean value was recorded.
Culm length, internode length and panicle length
The culm length of plant was measured from the ground level to panicle neck using regular ruler and panicle length was also measured from the same plant starting from panicle neck to the tip of panicle. Moreover, internode length was separately measured from six different plants.
Number of primary and secondary branches per panicle
The number of primary and secondary branch per panicle was counted for each individual sample plants for both wild-type and lup mutant.
Spikelet distance and fertility
Spikelet distance was separately measured for spikelet on first three, middle four and last three primary branches per panicle for both lup mutant and wild-type plants. Measurement was taken from six randomly selected plants. Finally, the total number of spikelet per panicle was determined and spikelet fertility percentage was calculated based the ratio of the number fully matured grains per panicle to total number of spikelet per panicle for lup mutant and wild-type plants.
Grain size and grain weight
Harvested grain was air-dried and stored at room temperature before measuring. Ten randomly selected, completely matured grains derived from mutant and wild-type were used to measure grain length (GL), grain width (GW) and grain shape (GS). The GL and GW were measured using digital microscope and measuring software. The GS was calculated as grain length divided by grain width. Grain weight was measured in grams as the weight of 1,000 fully ripened (14% moisture) grains per plant.
Genetic mapping of the lup locus
For genetic mapping, two F
2 mapping population from the cross between (1)
lup mutant and Milyang 23 (M.23), and (2)
lup mutant and Hwacheongbyo were used. The bulk segregant analysis (BSA) was used to identify the candidate markers linked to the
lup locus. Four mutant and four wild-type plants based on their phenotype were selected from
lup/M.23 F
2 population. A total of 91 Sequence-Tagged Sites (STS) markers of known chromosomal position throughout 12 chromosomes which were designed by Crop Molecular Breeding lab (CMB) were selected for BSA approach. Subsequently, phenotype and genotype of F
2 mapping population were used to determine gene position on the chromosome and to conduct fine mapping. Additional STS markers for fine mapping were developed based on the differences in sequence between the
japonica and
indica subsepecies (
Table 1).
Genomic DNA was extracted from leaf tissues using the CTAB method (
Chen and Ronald 1999). The PCR mixture was made with 1 μl DNA (30 ng/μl), 0.4 μl primers (10 μmol/μl), 2 μl 10× buffer, 0.4 μl dNTP (10 mmol/L), 0.3 μl Taq (5 U/μl) and 15.9 μl double-distilled water. PCR reaction system was performed at predenaturation temperature of 95°C for 5 min followed by 35 cycles of denaturation at 95°C for 30 second; annealing at 52–60°C depending on individual markers for 30 second; extension at 72°C for 40 second; with a final extension step at 72°C for 10 min. The PCR products were separated on 3.0% agarose gels which was stained with ethidium bromide and run in 0.5X TBE buffer and photographed.
Genomic DNA was extracted from young leaves of F2 population (cross between mutant and wild-type) by using CTAB method. The extracted DNA was pooled from 20 individuals (>5 μg) which had shown clear mutant phenotype. For Single Nucleotide Polymorphism (SNP) analysis, the bulked genomic DNA was sequenced using Illumina HiSeq2500 platform and the raw data of whole genome sequence of Hwacheongbyeo was also used. DNA libraries for Illumine HiSeq2500 platform was constructed using the kits (Truseq Nano DNA LT sample preparation kit, FC-121-4001). The quantitative Polymerase Chain Reaction (qPCR) was conducted using these libraries and then amplified clonal clusters were generated and performed pair end sequencing using Illumine HiSeq2500 (20 cycles). Base calling was carried out by instrument software Real Time Analysis (RTA).
Alignment of the of short reads to reference sequences and SNP and InDel calling
Whole genome sequencing and SNP analysis were performed to identify the mutated genomic region. The pre-processing of the whole paired end short reads from F
2 bulk DNA was carried out using btrim0.2.0 (
http:graphics.med.yale.edu/trim). The cut off thresholds for low quality base were phred-score 20 and minimum read length 50. Processed short read were mapped and aligned to publicly available Nipponbare genome sequence (build 5 version;
http:rgp.affrc.go.jp/E/IRGSP/Build5/buil5.html) with clc_ref_assemble (V 4.2104315).
The whole genome sequence for Hwacheongbyeo was also filtered and mapped to Nipponbare followed by same procedures. Subsequently, SNP detection and SNP calling were performed by find variations (V 4.21.10435). SNP filtering options are read depth of the site ≥5, variant frequency of major base ≥0.9. After mapping and SNP calling, causal SNPs and InDels involved in the phenotype of mutant were detected by following modified MutMap methods (
Abe et al. 2012): (1) Remove the redundant SNPs and InDels between wild-type and mutant from the list, (2) Calculate SNP index, (3) Pick SNPs and InDels (SNP index=1) located in annotated genes in the candidate region previously revealed by fine mapping. (4) Remove those SNPs located outside the intragenic region, and (5) Remove those SNPs causing a synonymous mutation. Finally those Homozygous SNPs which were specific to F
2 mutant bulk were identified as causal mutation points. SNP index is the ratio between the number of reads of a mutant SNP and the total number of reads at the each position.
RESULTS
Agronomic trait analysis
The mean values of agronomic traits analysed is displayed in
Table 2. Agronomic traits between
lup mutant and wild-type showed significant differences, especially on panicle structure after heading (
Fig. 1a). Abortion of spikelet on tip of the first primary branch started to be shown up immediately after heading of panicle. The distance between spikelet on the primary branch was significantly elongated in
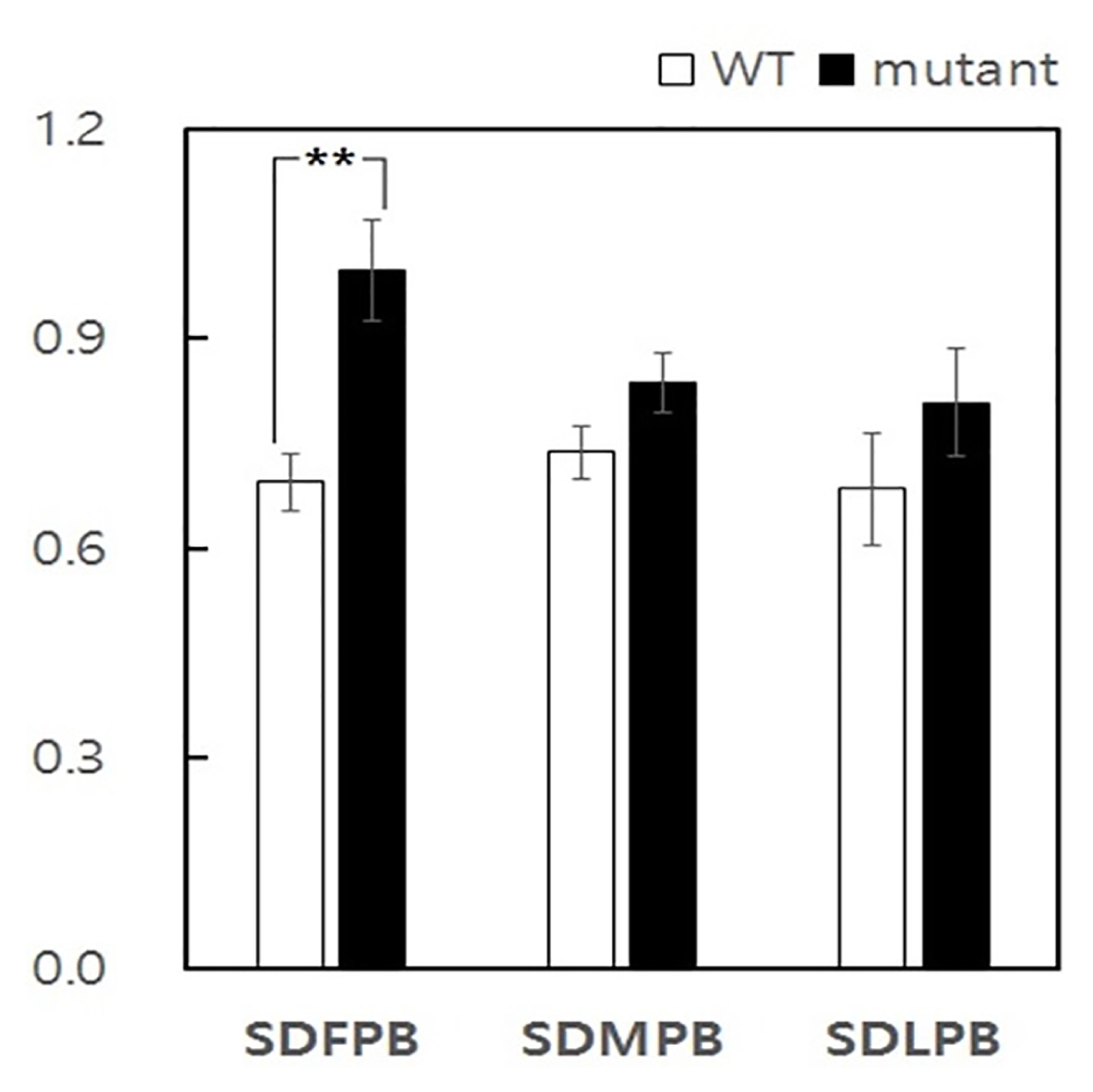
lup mutant compared to wild-type. The spikelet distance of the first three primary branches, four middle primary branches and last three primary branches were analysed (
Fig. 2). The result of this analysis indicated spikelet distance on first primary branch in
lup mutant was significantly different with that of middle and lower primary branch spikelet distance of wild-type. Accordingly, the deformity of spikelet distance induced due to mutation was specifically confined to the first three primary branches of mutant plants.
Although the number of secondary branches and spikelet per secondary branches did not show any significant differences, the number of primary branches and spikelet per primary branches was significantly reduced compared to wild-type. Hence, the total number of spikelet per panicle in
lup mutant was significantly reduced compared to wild-type. However, the length of the primary and secondary branches were not significantly different between
lup mutant and wild-type (
Table 2).
Other agronomic traits also showed significant difference between
lup mutant and wild-type. Culm length (the average of culm length from the base to the neck node), panicle length (length from the neck node to the panicle tip), and tiller number were significantly reduced by 2.06 cm, 2 cm and 2.5 respectively compared to wild-type. Grain length and width was also significantly reduced by 0.508 mm and 0.464 mm in
lup mutant, respectively (
Fig. 1e and
Table 2). In consistent with this result, the 1,000-grain weight of the
lup mutant was significantly reduced compared to wild-type, despite the
lup mutant displayed stay-green even after heading (
Fig. 1c). However, there was no significant change in grain shape, which was calculated as the ratio of grain length to width. Moreover, the translucent nature of wild-type seeds were observed to be changed to chalky in
lup mutant (
Fig. 1e).
We developed F
2 populations from the crosses between (1)
lup mutant and Hwacheongbyeo (
japonica), and (2)
lup mutant and Milyang 23 (a tongil-type cultivar from
indica/
japonica cross). All F
1 exhibited wild-type phenotype. Out of 262 plants in F
2 population, 209 plants showed an appearance of wild-type while 53 plants were similar as the
lup mutant plants. The ratio of this segregation was 209:53 which correspond with 3:1 Mendelian ratio. Similar result was obtained from the F
2 population of the cross between
lup mutant and M.23 (
Table 3). These results therefore indicated that a single recessive gene, which was tentatively designed as
lup, was involved in the phenotypic alteration of
lup mutant.
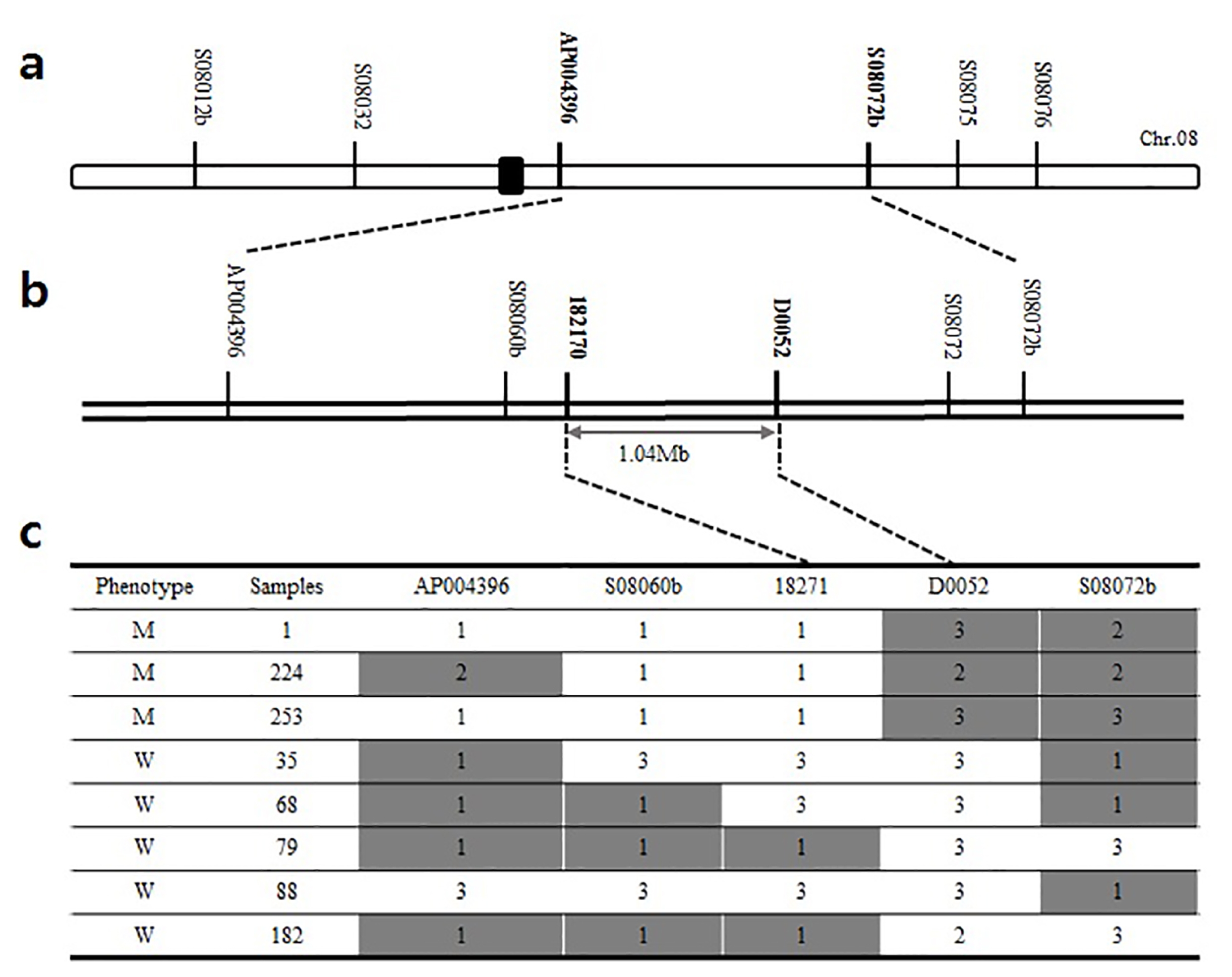
To investigate the genomic location of
lup gene, fine mapping was conducted using the F
2 population generated from the cross combination of
lup mutant (
japonica-type) and M.23. Bulk Segregant Analysis (BSA) was carried out with 91 polymorphic STS primers that evenly distributed throughout 12 rice chromosomes. Consequently, the
lup locus was mapped in the long arm of chromosome 8 between two STS markers, AP004396 and S08072b (
Fig. 3a). Additional STS markers between these two flanking markers were designed based on the difference in DNA sequence between
indica and
japonica varieties from databases (
http://www.ncbi.nlm.nih.gov/ and
http://rgp.dna.affrc.go.jp/). Further fine mapping was made and the
lup gene region was narrow downed up to 1.04 Mb using an additional STS markers; 18217 and D0052 (
Fig. 3b). Subsequently, modified MutMap analysis was performed to identify candidate genes.
The genomic DNA samples isolated from 20 individual
lup mutants in segregating F
2 population (cross between Hwacheongbyeo and
lup mutant) were pooled and used for whole genome sequencing with depth of more than 10x coverage. To identify candidate genes and causal SNP position, the SNP analysis was performed with the whole-genome sequencing data of Hwacheongbyeo and the bulk of F
2 individuals displaying the mutant phenotype. The whole genome sequence of
lup mutant and Hwacheongbyo were aligned to the reference sequence and those SNPs and InDels found to be redundant between
lup mutants and wild-type were removed from the lists for they were not accounting for the expressed mutant phenotypic characters. Subsequently, SNPs and InDels found outside the intragenic region were also removed. Finally, we found a total of 348 SNPs within the candidate
lup gene region after removing those synonymous mutation points associated with 51 candidate genes (
Table 4).
DISCUSSION
Panicle structure is one of the most important agronomic traits that critically determine the yielding potential of rice. The basic unit of the rice panicle which finally is described as yield is known to be the total number of fertile spikelet per panicle. The abortion of spikelet usually causes degeneration of floral organs, leading to reduction of rice yield. Spikelet degeneration can occur from the basal or apical end of the panicle and lead to defective, whitish spikelet, and eventually fewer spikelet per panicle (
Li et al. 2009;
Cheng et al. 2011).
The
lup mutant has also been observed to cause significant reduction to yield related components (
Table 2). Most of the agronomic traits were reduced in
lup mutant compared to wild-type. Traits in the panicle were the key indicator for
lup mutant which identified from wild-type. The elongated spikelet distance on the first primary branch was clearly observed during initial panicle exsertion for identifying mutant plant from normal one. Subsequently, soon after panicle exsertion, those spikelet found on tip of first three primary branches tend to be bleached gradually and be aborted for more clear identification of mutant plants from F
2 population.
Mutant plants had maintained greenness even after attaining physiological maturity. However, lup mutant was found to have less number of total spikelet and grain weight. Even though length of primary and secondary branch, number of secondary branch and spikelet on secondary branch did not show significant difference compared to wild-type, the difference observed in total number of spikelet per panicle was significant due to reduced number of primary branch and spikelet on primary branch.
From the result of comparison of the spikelet distance on first, middle and last primary branch of mutant and wild-type, it becomes evident that the first three primary branches were most affected by lup gene. The comparison of spikelet distance through three region between mutant and wild-type revealed significant difference only in the first three primary branches.
The overall results of agronomic trait analysis indicated that lup mutant had shown considerable reduction in spikelet fertility and grain related traits. More importantly, mutant exhibited comprehensive reduction on the characters of the first primary branch which mainly accounted for overall reduction. Therefore, it could be concluded that lup gene plays key role in determining the yielding potential of rice.
Understanding the genetic mechanism underlying the inflorescence architecture is practically important for developing high yield varieties in cereal breeding programs. Several genes including
SP1,
APO1,
MOC1,
LAX,
RCN1,
FZP,
FON1, and
FON4 have been identified as being associated with panicle architecture in rice and as being responsible for causing yield loss (
Nakagawa et al. 2002; Komatsu
et al. 2003;
Li et al. 2003,
2009;
Ashikari et al. 2005;
Chu et al. 2006;
Moon et al. 2006). Most of the identified genes affecting panicle architecture are involved in the basal degeneration of spikelet and/or rachis branches. However, the
lup mutant in this study has exhibited unique morphological characteristics which has been observed only on the first primary branch. Overall agronomic trait analysis indicated that
lup is strongly associated with panicle related traits. Therefore, identification of a causal mutation points has high priority for further analyses to understand the biological role of
lup gene in panicle development.
In this study, we had tried to identify mutation points using F
2 bulk sequenced data and genetic mapping results. Slightly modified MutMap method was used for analysing SNPs in the sequenced data. The MutMap method in the source document (
Abe et al. 2012) was performed with various programs and several stapes to identify causal mutation points where SNP index is near 1. For our case, we slightly modified the approach and analysed the SNP data of the whole genome sequence and obtained 348 candidate mutation points which are included in 51 candidate genes in the flanking region. Among candidate genes, 23 were hypothetical genes. The remaining 28 genes were expressed proteins with specific description mentioned in
Table 4.
In conclusion, the result of agronomic trait analysis indicated that lup has been involved in different agronomic traits particularly related to the panicle traits. With the genetic analysis and fine mapping, lup locus was mapped to the long arm of chromosome 8. Further analysis by modified MutMap revealed that there were 348 casual mutation points in 51 candidate genes in the flanking region. Further researches, including cloning and the functional analysis of LUP, may interpret the mechanism of the phenotypes observed in the lup mutant, and provide further knowledge of panicle development in rice.
ACKNOWLEDGEMENTS
This work was supported by a grant from Next-Generation BioGreen 21 program (Plant Molecular Breeding Center No. PJ01102401), Rural development Administration, Republic of Korea.
Fig. 1Comparison of the agronomic traits between wild-type and lup mutant. (a) Panicle structure, (b) Plants before heading, (c) after heading, (d) Internode, and (e) grain size. Scale bars (a) 3 cm, (b–c) 20 cm, (d) 5 cm, and (e) 5 mm.

Fig. 2Spikelet distance (SD) variability between lup mutant and WT primary branches. SDFPB Spikelet distance of first primary branch, SDMPB Spikelet distance on middle primary branches, and SDLPB Spikelet distance on last primary branch. **Significant at 1% level of probability.
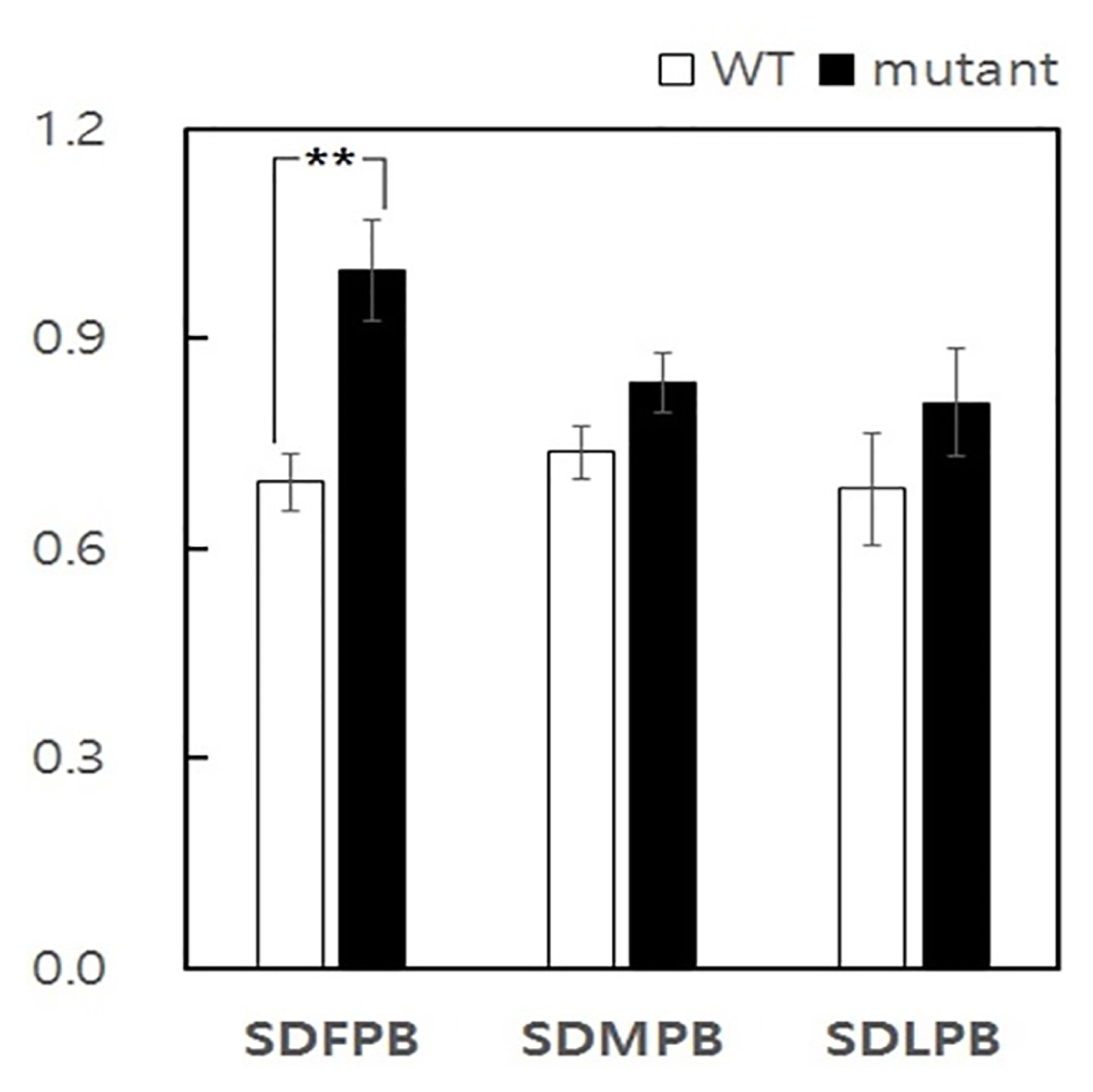
Fig. 3Genetic map of lup mutant: (a) Primary mapping of the lup locus on chromosome 8, (b) the lup locus was fine mapped with the adjacent markers. (c) Graphical genotype results of fine mapping. W: wild type, M: mutant type, 1: mutant type, 2: wild type, 3: hetero type.
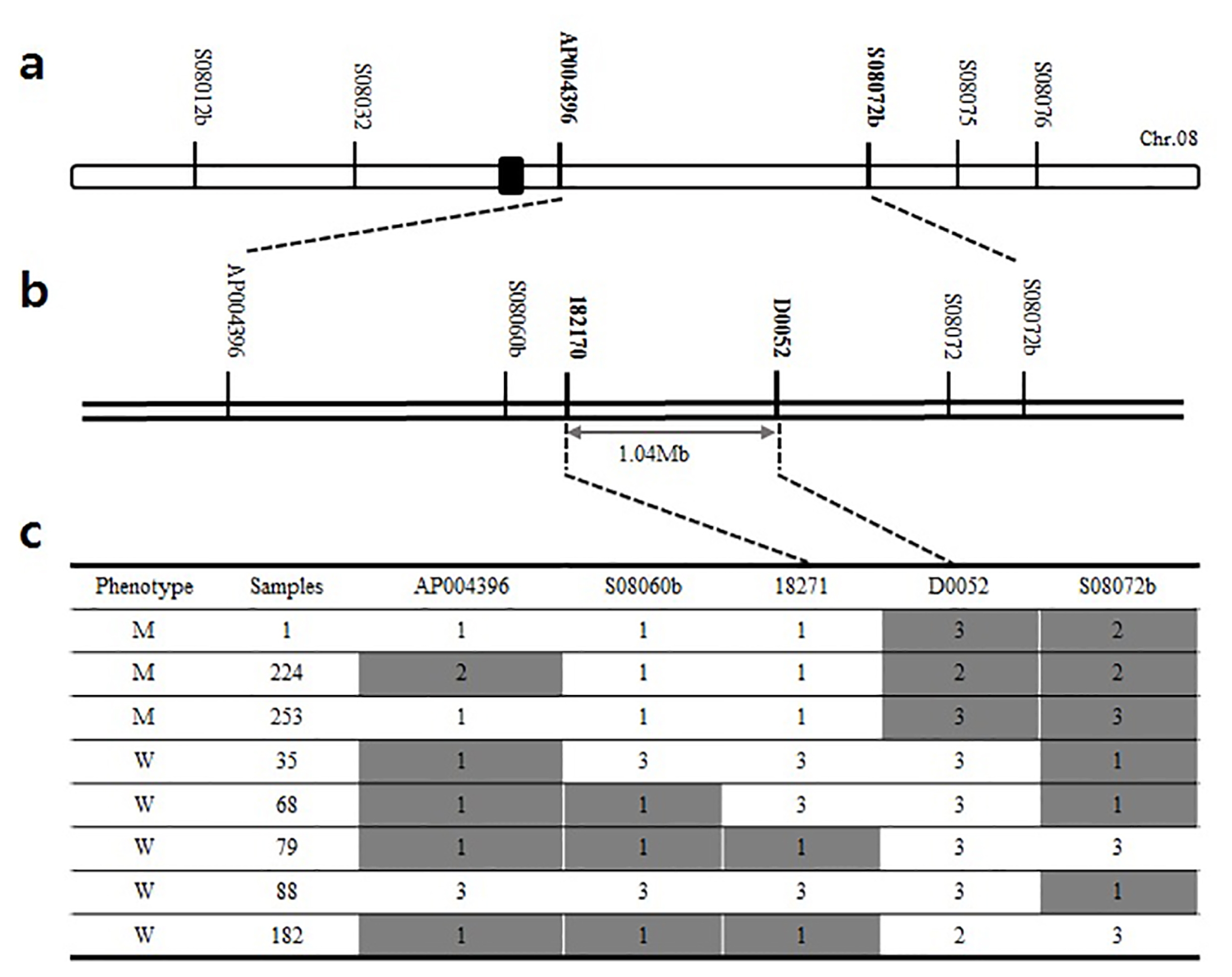
Table 1The PCR-based molecular markers used in fine mapping of the lup gene.
Table 1
|
STS marker |
Forward primer sequence (5′-3′) |
Reverse primer sequence (5′-3′) |
Size (bp) |
|
S08012b |
GAAGCCAAAATGGGAGAGAA |
TTTTGTGCTGCCACTGAAAG |
224 |
|
S08032 |
GTACGAGAACCTGCAGAGCA |
TTTCCACCTCAACCAAGTCC |
208 |
|
AP004396 |
TACAAGGATGCGGAAAGACC |
CCACCATTAAAGCAGCACAA |
246 |
|
S08060b |
GGAGAGGAAAGTGGGAGAGG |
ACCTCGTGATTGGTTGCTTC |
223 |
|
182170 |
CAAAGCTGGGTGTAGGGAGA |
CTGGTCAAGTTGAGGCACAG |
263 |
|
S08070 |
ACTACATCGTCCGGCTCAAG |
GGTGCAAGCAAGCAATGAT |
143 |
|
S08072b |
AAATTAATGGATGTACTCTTCTGTTTG |
CAGCAGCGGATTTGCTATC |
265 |
|
S08075 |
AAACAAGTTTGCGGACCAAG |
TGAGTGATAAACAACTCACAACAAAA |
256 |
|
S08076 |
TTGCATGTGTCGTCTGTCCT |
GTTTCAAGCAGTTGGGAAGC |
212 |
Table 2Agronomic traits of the lup mutant and wild-type.
Table 2
|
Traits |
PBL (cm) |
SBL (cm) |
PB |
SB |
SPP |
SPS |
TSP |
SPF (%) |
1000-GW (g) |
CL (cm) |
PL (cm) |
TN |
GL (mm) |
GW (mm) |
GS (mm) |
|
WT |
6.8 |
1.49 |
9.75 |
13.75 |
57.50 |
37.88 |
95.37 |
94.21 |
26.30 |
101.50 |
18.86 |
11.5 |
9.14 |
5.18 |
1.77 |
|
lup
|
7.3 |
1.46 |
9.20 |
13.90 |
49.28 |
37.38 |
86.67 |
66.79 |
18.67 |
89.50 |
16.80 |
9 |
8.64 |
4.71 |
1.84 |
|
Difference |
NS |
NS |
*
|
NS |
**
|
NS |
*
|
**
|
*
|
**
|
**
|
*
|
*
|
*
|
NS |
Table 3Genetic segregation of lup mutant in two F2 populations.
Table 3
|
Cross combination |
Wild type |
Mutant |
Total |
χ2 (3:1) |
|
lup mutant / Milyang 23 |
357 |
131 |
488 |
0.885 |
|
lup mutant / Hwacheongbyeo (WT) |
209 |
53 |
262 |
1.92 |
Table 4Candidate genes comprising SNPs in the lup mutant.
Table 4
|
No. |
Gene name |
No. of SNPs |
Nucleotide length |
Description |
|
1 |
Os08g0384500 |
3 |
4161 |
Similar to PDR-like ABC transporter (PDR3 ABC transporter). |
|
2 |
Os08gt0384602 |
1 |
2755 |
Hypothetical gene. |
|
3 |
Os08g0384700 |
3 |
2953 |
Hypothetical gene. |
|
4 |
Os08g0384900 |
1 |
4269 |
Zinc finger, RING/FYVE/PHD-type domain containing protein. |
|
5 |
Os08g0385000 |
1 |
455 |
Conserved hypothetical protein. |
|
6 |
Os08g0385400 |
1 |
3466 |
Conserved hypothetical protein. |
|
7 |
Os08g0385900 |
1 |
6297 |
Similar to Transformer-2-like protein. |
|
8 |
Os08g0386700 |
4 |
5856 |
Similar to Avr9 elicitor response protein-like. |
|
9 |
Os08g0386800 |
1 |
907 |
Hypothetical conserved gene. |
|
10 |
Os08g0386900 |
1 |
2635 |
Prefoldin domain containing protein. |
|
11 |
Os08g0387050 |
7 |
2867 |
Hypothetical conserved gene. |
|
12 |
Os08g0387200 |
8 |
7262 |
Protein of unknown function DUF81 family protein. |
|
13 |
Os08g0388300 |
1 |
13132 |
Hypothetical conserved gene. |
|
14 |
Os08g0388900 |
1 |
7795 |
Similar to para-hydroxybenzoate--polyprenyltransferase. |
|
15 |
Os08g0392100 |
1 |
1381 |
2OG-Fe(II) oxygenase domain containing protein. |
|
16 |
Os08g0392300 |
25 |
2237 |
Similar to Oxidoreductase, 2OG-Fe oxygenase family protein, expressed. |
|
17 |
Os08g0392500 |
25 |
10744 |
Hypothetical conserved gene. |
|
18 |
Os08g0392700 |
8 |
749 |
Conserved hypothetical protein. |
|
19 |
Os08g0393600 |
4 |
2474 |
Protein-tyrosine phosphatase-like, PTPLA domain containing protein. |
|
20 |
Os08g0393700 |
4 |
1332 |
Similar to predicted protein. |
|
21 |
Os08g0393900 |
3 |
640 |
Hypothetical gene. |
|
22 |
Os08g0394000 |
6 |
758 |
Conserved hypothetical protein. |
|
23 |
Os08g0395300 |
1 |
1826 |
Similar to AP-4 complex subunit sigma-1. |
|
24 |
Os08g0395700 |
1 |
1394 |
Conserved hypothetical protein. |
|
25 |
Os08g0395800 |
2 |
1937 |
Protein of unknown function DUF247, plant family protein. |
|
26 |
Os08g0396200 |
6 |
698 |
Hypothetical gene. |
|
27 |
Os08g0396500 |
7 |
3203 |
Hypothetical conserved gene. |
|
28 |
Os08g0396700 |
47 |
13559 |
D1 domain containing protein. |
|
29 |
Os08g0397800 |
6 |
770 |
Conserved hypothetical protein. |
|
30 |
Os08g0397900 |
3 |
2682 |
Similar to Holocarboxylase synthetase. |
|
31 |
Os08g0398000 |
9 |
6202 |
ABC transporter-like domain containing protein. |
|
32 |
Os08g0398300 |
3 |
5664 |
ABC transporter-like domain containing protein. |
|
33 |
Os08g0398350 |
73 |
6346 |
Similar to ABC transporter family, cholesterol/phospholipid flippase. |
|
34 |
Os08g0398400 |
24 |
4030 |
Similar to Hypersensitive-induced response protein. |
|
35 |
Os08g0398800 |
2 |
1703 |
Cleavage and polyadenylation specificity factor, 25 kDa subunit domain containing protein. |
|
36 |
Os08g0398966 |
2 |
507 |
Hypothetical gene. |
|
37 |
Os08g0399300 |
2 |
2618 |
Hypothetical conserved gene. |
|
38 |
Os08g0399550 |
3 |
1436 |
Hypothetical conserved gene. |
|
39 |
Os08g0399600 |
1 |
1501 |
Similar to Elongation factor Ts. |
|
40 |
Os08g0399900 |
6 |
1498 |
Conserved hypothetical protein. |
|
41 |
Os08g0400000 |
2 |
5349 |
Similar to Puromycin-sensitive aminopeptidase (EC 3.4.11.-) (PSA). |
|
42 |
Os08g0400200 |
1 |
3653 |
Similar to SET domain protein. |
|
43 |
Os08g0401100 |
1 |
3001 |
Ankyrin repeat containing protein. |
|
44 |
Os08g0401601 |
4 |
1958 |
Hypothetical genes. |
|
45 |
Os08g0401800 |
14 |
2568 |
Similar to C2; Peptidase, cysteine peptidase active site. |
|
46 |
Os08g0402001 |
4 |
653 |
Hypothetical conserved gene. |
|
47 |
Os08g0402100 |
2 |
1517 |
Lateral organ boundaries, LOB domain containing protein. |
|
48 |
Os08g0402500 |
2 |
1151 |
Conserved hypothetical protein. |
|
49 |
Os08g0402600 |
1 |
1978 |
Pentatricopeptide repeat domain containing protein. |
|
50 |
Os08g0402700 |
7 |
4088 |
Similar to 10A19I.7. |
|
51 |
Os08g0402800 |
2 |
5457 |
Protein of unknown function DUF6, transmembrane domain containing protein. |
References
- Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R. 2012. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol. 30: 174-179.
- Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M. 2005. Cytokinin oxidase regulates rice grain production. Science. 309: 741-745.
- Bai J, Zhu X, Wang Q, Zhang J, Chen H, Dong G, Zhu L, Zheng H, Xie Q, Nian J, Chen F, Fu Y, Qian Q, Zuo J. 2015. Rice TUTOU1 Encodes a SCAR-like Protein that Is Important for Actin Organization and Panicle Development. Plant Physiol. 169: 1179-1191.
- Cassman KG, Dobermann A, Walters DT, Yang H. 2003. Meeting cereal demand while protecting natural resources and improving environmental quality. Annu Rev Environ Resour. 28: 315-358.
- Chen DH, Ronald PC. 1999. A rapid DNA minipreparation method suitable for AFLP and other PCR applications. Plant Mol Biol Rep. 17: 53-57.
- Cheng Z, Liu C, Yang D, Lu L, Ye L, Zheng X, Ye X. 2011. Molecular markers-assisted selection of restorer line of dense and erect panicle. Mol Plant Breed. 9: 561-566.
- Chu H, Qian Q, Liang W, Yin C, Tan H, Yao X, Yuan Z, Yang J, Huang H, Luo D, Ma H, Zhang D. 2006. The floral organ number4 gene encoding a putative ortholog of Arabidopsis CLAVATA3 regulates apical meristem size in rice. Plant Physiol. 142: 1039-1052.
- He G, Zhu X, Elling AA, Chen L, Wang X, Guo L, Deng XW. 2010. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 22: 17-33.
- Huang X, Qian Q, Liu Z, Sun H, He S, Luo D, Xia G, Chu C, Li J, Fu X. 2009. Natural variation at the DEP1 locus enhances grain yield in rice. Nat Genet. 41: 494-497.
- Jiao Y, Wang Y, Xue D, Wang J, Yan M, Liu G, Dong G, Zeng D, Lu Z, Zhu X, Qian Q, Li J. 2010. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet. 42: 541-544.
- Khush G. 2005. What it will take to Feed 5.0 Billion Rice consumers in 2030. Plant Mol Biol. 59: 1-6.
- Komatsu M, Maekawa M, Shimamoto K, Kyozuka J. 2001. The LAX1 and FRIZZYPANICLE 2 genes determine the inflorescence architecture of rice by controlling rachis-branch and spikelet development. Dev Biol. 231: 364-373.
- Li S, Qian Q, Fu Z, Zeng D, Meng X, Kyozuka J, Maekawa M, Zhu X, Zhang J, Li J, Wang Y. 2009. Short panicle1 encodes a putative PTR family transporter and determines rice panicle size. Plant J. 58: 592-605.
- Li S, Zhao B, Yuan D, Duan M, Qian Q, Tang L, Wang B, Liu Xg, Zhang J, Wang J, Sun J, Liu Z, Feng YQ, Yuan L, Li Ch. 2013. Rice zinc finger protein DST enhances grain production through controlling Gn1a/OsCKX2 expression. Proc Natl Acad Sci. 110: 3167-3172.
- Li X, Qian Q, Fu Z, Wang Y, Xiong G, Zeng D, Wang X, Liu X, Teng S, Hiroshi F, Yuan M, Luo D, Han B, Li J. 2003. Control of tillering in rice. Nature. 422: 618-621.
- Moon S, Jung KH, Lee DE, Lee DY, Lee J, An K, Kang HG, An G. 2006. The rice FON1 gene controls vegetative and reproductive development by regulating shoot apical meristem size. Mol Cells. 21: 147-152.
- Nakagawa M, Shimamoto K, Kyozuka J. 2002. Overexpression of RCN1 and RCN2, rice TERMINAL FLOWER 1/CENTRORADIALIS homologs, confers delay of phase transition and altered panicle morphology in rice. Plant J. 29: 743-750.
- Oikawa T, Kyozuka J. 2009. Two-step regulation of LAX PANICLE1 protein accumulation in axillary meristem formation in rice. Plant Cell. 21: 1095-1108.
- Sakamoto T, Matsuoka M. 2004. Generating high-yielding varieties by genetic manipulation of plant architecture. Curr Opin Biotech. 15: 144-147.
- Terao T, Nagata K, Morino K, Hirose T. 2010. A gene controlling the number of primary rachis branches also controls the vascular bundle formation and hence is responsible to increase the harvest index and grain yield in rice. Theor Appl Genet. 120: 875-893.
- Wang Y, Li J. 2008. Molecular basis of plant architecture. Annu Rev Plant Biol. 59: 253-279.
- Yoshida , Nagato . 2011. Flower development in rice. J Exp Bot. 62: 4719-30.
- Youlin P, Zhenyu G, Bin Z, Chaolei L, Jie X, Banpu R, Jiang H, Guojun D, Longbiao G, Guohua L, Qian Q. 2014. Fine mapping and candidate gene analysis of a major QTL for panicle structure in rice. Plant Cell Rep. 33: 1843-1850.